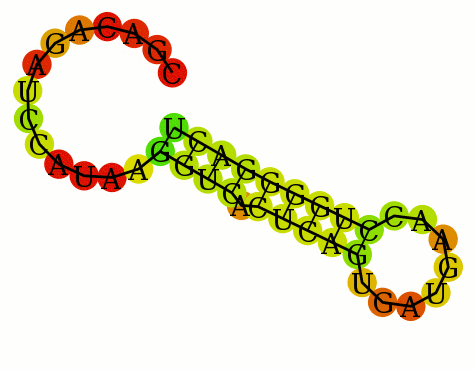
1 CGACAGAUCCAUAAGGUCACUCAGUGAUGAACCUGGGGACU 1 ..............((((.(((((........)))))))))
Reducing the number of threads (using -T 1) speeds up cuda RNAfold by about 18%.
RNAfold < jabbari_1.lis mfe: -9.80 kcal/mol Suboptimal range: [-9.80 kcal/mol - -8.82 kcal/mol] CGACAGAUCCAUAAGGUCACUCAGUGAUGAACCUGGGGACU ..............((((.(((((........))))))))) (-8.90) .......(((...((((...(((....)))))))..))).. (-9.30) ....((.(((...((((...(((....)))))))..))))) (-9.80) ....((..((...((((...(((....))))))).))..)) (-8.90) ....((.(((...((((...(((....))))))).))).)) (-9.00) 0.005u 0.006s 0:00.11 0.0% 0+0k 0+0io 0pf+0wNote Jabbari says the folding is
structure in thesis: ..............((((.(((((........))))))))) Mirela's Simfold energy: -10.10 ADP Simfold energy: -10.100001 Approximate ADP timing: 4 sec difference: NoneRNAfold webserver says:
1 CGACAGAUCCAUAAGGUCACUCAGUGAUGAACCUGGGGACU 1 ..............((((.(((((........)))))))))
Create random sequences. Eg gawk -f rand_dna.awk -v "total=1000" /dev/null > t1000.fasta
cudaMemcpyToSymbol calls in cudalib.cu have also been tweaked.
Delete RNAfold and rebuild it using make
/usr/lib/gcc/x86_64-redhat-linux/4.1.2/../../../../lib64/libreadline.so: undefined reference to `PC' /usr/lib/gcc/x86_64-redhat-linux/4.1.2/../../../../lib64/libreadline.so: undefined reference to `tgetflag' /usr/lib/gcc/x86_64-redhat-linux/4.1.2/../../../../lib64/libreadline.so: undefined reference to `tgetent' /usr/lib/gcc/x86_64-redhat-linux/4.1.2/../../../../lib64/libreadline.so: undefined reference to `UP' /usr/lib/gcc/x86_64-redhat-linux/4.1.2/../../../../lib64/libreadline.so: undefined reference to `tputs' /usr/lib/gcc/x86_64-redhat-linux/4.1.2/../../../../lib64/libreadline.so: undefined reference to `tgoto' /usr/lib/gcc/x86_64-redhat-linux/4.1.2/../../../../lib64/libreadline.so: undefined reference to `tgetnum' /usr/lib/gcc/x86_64-redhat-linux/4.1.2/../../../../lib64/libreadline.so: undefined reference to `BC' /usr/lib/gcc/x86_64-redhat-linux/4.1.2/../../../../lib64/libreadline.so: undefined reference to `tgetstr' collect2: ld returned 1 exit status
It appears this is a known problem with centos and Red hat linux distributions.
gcc RNAfold.o adplib.o rnalib.o RNAfold_mfe.o -lreadline -lm -lncurses -o RNAfold
This is compounded by the fact that RNAfold's use of memory continues to grow as it outputs this huge file. (It looks like memory is allocated by myalloc() but not freed by memory_free().)
./RNAfold -e 0 < input_filewill ensure only the best folds are output.